Some Latest & Important Post
Recent Posts
13,Dec 2019 Mir Shahroz
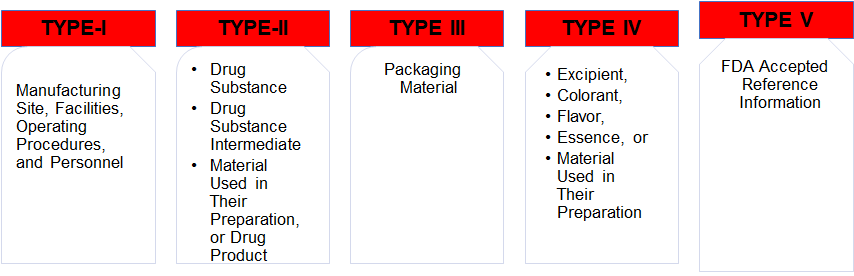
DRUG MASTER FILE (US-FDA)
DRUG MASTER FILE (US-FDA SUBMISSION)
18,Nov 2019 Mir Shahroz
POST APPROVAL
Once approval has been obtained, there is usually a need for variations on the original marketing authorisation
19,Nov 2019 Mir Shahroz
Medical Devices
Classification of medical device, preparation of technical master file, Clinical and non-clical expert report of medical device and in vitro diagnostic.

18,Nov 2019 Mir Shahroz
DUE DILIGENCE
Are you contemplating a licensing deal, a strategic partnership or a merger & acquisition
15,Dec 2019 Mir Shahroz
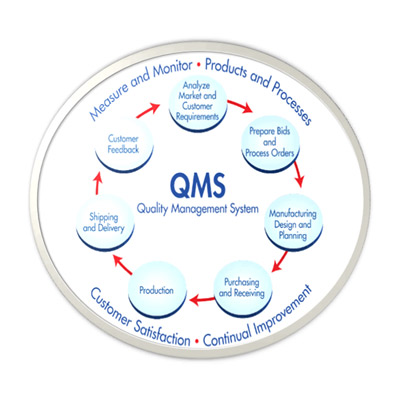
QMS/cGMP Audit Preparation and Establishment
QMS/cGMP Audit Preparation and Establishment
for Pharmaceutical/Neutraceuticals/Herbal/Medical device/Excipients/Drug Substance company
GAP analysis, System establishment, Facilitation of Regulatory Agency Audit, Deficiency response (Agency) etc,
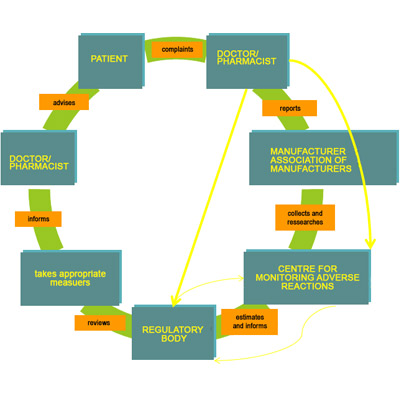
11,Dec 2019 Mir Shahroz
PHARMACOVIGILANCE
Pharmacovigilance system (INDIA)